
Quickguide on NMR calculation
Published:
This document introduces a fundamental workflow for performing NMR calculations on small organic molecules. It aims to help experimentalists or newcomers to computational chemistry quickly gain hands-on experience with NMR calculations. For a more detailed and comprehensive content, please refer to this chapter of computational NMR.
Introduction
Nuclear Magnetic Resonance (NMR) spectroscopy is a powerful analytical tool in chemistry, providing invaluable information about the structure and dynamics of molecules. It plays a crucial role in organic chemistry, allowing researchers to confirm the identity and purity of synthesized compounds. However, interpreting NMR spectra can sometimes be challenging, especially for complex molecules or those with overlapping signals. In such cases, computational NMR calculations can provide valuable support for structure verification.
Computational Chemistry and NMR
Computational chemistry has established itself as a complementary approach to experimental techniques, offering insights into molecular properties and behavior. In the context of NMR, computational methods can predict chemical shifts, coupling constants, and spin-spin interactions, aiding in the interpretation of experimental spectra. The most commonly used methods for NMR calculations are based on quantum mechanics, such as Density Functional Theory (DFT) and ab initio methods.
However, these calculations do not directly provide the chemical shifts observed in the spectrum. Instead, they compute the shielding tensor, which must be converted to the chemical shift by calculating the relative value to a reference compound, such as tetramethylsilane (TMS). An alternative approach is to use empirical scaling factors to obtain the chemical shifts. For simple organic molecule, CHESHIRE is a databased to go for this perpose.
Workflow for NMR Calculations
Before you start
Before starting any calculation or simulation, it is crucial to consider the bigger picture of the entire process and determine the best approach for molecular modeling. Several key factors should be taken into account to ensure that the calculations are affordable, practical, and chemically accurate. This will significantly reduce the likelihood of encountering discrepancies between computational results and experimental data due to unreliable computational methods.
Computational Resources: Assess the available computational resources to ensure that the chosen computational methods are feasible and efficient.
Molecular Size and Complexity: Consider the size and complexity of the molecule being studied. Larger molecules or those with many atoms and intricate geometries/electronic structure may require more advanced computational methods.
Protonation and Deprotonation States: Evaluate the possible protonation and deprotonation states of the molecule, especially if it is a zwitterion. The protonation state can significantly influence the molecular geometry, electronic structure, and NMR properties. Accurately represent the protonation state of the molecule in the calculations to ensure reliable results.
Solvent Effects: Take into account the solvent used in the NMR experiment, as it can have a substantial impact on the molecular geometry, electronic structure, and NMR properties. Incorporating solvent effects in the calculations, either implicitly or explicitly, can improve the agreement between computational and experimental results.
By carefully considering these factors and selecting the most appropriate computational methods, researchers can optimize the reliability and accuracy of their NMR calculations, minimizing the risk of discrepancies between computational and experimental results.
Conformational searching
The first step is comformational searching. after you construct the molecule with any software that you are comfortable with (i.e. GaussView), it is very important to explore the potential structure with the molecule. Becuase that based on Boltzmann equation, we know that the ratio of two conformation of the same molecule depends on the relative free energies of them. (This website is very helpful, type in relative energies and it will give the boltzmann population ratio at room temperature). A conformer that is 3 kcal/mol higher in energy has less than 1% weight compared to the “better” conformation. Since the computed NMR is very sensitive to the geometry of a molecule, we need to confirm that we have the structure that weights, or the one that has the lowest energy.
- How? Right now the most prevalent/reliable tool for running conformational searching is
xTB-CREST, which runs a metadynamics simulation by adding potential into the RMSD change of structures and therefore efficiently explore the potential energy surface using semi-empirical method. To installCREST, please follow the instruction fromCRESTdocumentation website here
Recently Grimme published the lastest version of CREST 3.0. This tutorial focus on the latest version of CREST that before 3.0 due to the reason of stability.
- commands to specify
We will not go deep into other specific metadynamics settings, for you interest you please check it here.
Once you have the .xyz file of your molecule and CREST is ready to run,:
crest struc.xyz --gfn2 --chrg 0 --ewin 7 --opt --v4 --alpb methanol > crest.out
- crest: The command to run the CREST program for conformational searching. molecule.xyz: The input file containing the coordinates of your molecule in XYZ format. Replace “molecule” with the actual name of your file.
- -gfn2: Specifies the GFN2 method for conformational searching.
- –chrg 0: Sets the charge of the molecule to 0. Adjust this value based on the actual charge of your molecule.
- –alpb water: Enables the ALPB (Analytical Linearized Poisson-Boltzmann) implicit solvation model with water as the solvent.
- -ewin 7: Sets the energy window to 7 kcal/mol. Conformers within this energy window will be considered during the search.
Make sure to replace molecule.xyz with the actual name of your XYZ file. Also, ensure that you have the necessary permissions and environment settings to run the crest command in your terminal or command prompt.
After the job is finished, you will see an output file named “crest_conformers.xyz”. To group all conformers into clusters (because there could be duplicate structures), it is convenient to use a script developed by Tian Lu to filter the structures by setting thresholds for structural similarity and energy differences. You may download the file from here (right click and then click Save Link As...). This file is extracted from a the Molclus package by Lu.
To run isostat, put it under the folder of your CREST working directory (where you have the crest_conformers.xyz!). Then give it permission and excute it.
chmod 755 isostat
./isostat
You will see it ask for the filename; simply type in crest_conformers.xyz and press enter. You may use its default settings for energy and structure thresholds by just hitting enter. Afterwards, you will see it try to group the structures in crest_conformers.xyz. Once it is done, you will see a long list of clustered structure names, their relative potential energies in kcal/mol, and the RMSD of the structures.
At the same time, you will see a file named cluster.xyz written out; this is the result of our conformational search and what we need for the following optimization. You may open the .xyz file with any visulization software, read the structures, and see how low-energy and high-energy conformations look (under the level of the semi-empirical method!).
Molecular geometry optimization
To obtain DFT level structures, we need to reoptimize the structures from cluster.xyz. Therefore, you need to save each of the structures in the cluster.xyz file and convert them into Gaussian optimization input files. If you are using the CHESHIRE scaling factors, pick one of the levels displayed in the tables. Find the solvent, get the table for that solvent, and select the scaling factors you want. The level of your computation must align with the methods on the table for geometry optimization and NMR calculations.
However, it is possible that you get 100 structures from cluster.xyz, and you don’t want to waste your computational time optimizing all of them using a very high level of theory. A more efficient and computationally economical approach is to optimize them with a lower level of theory, screen out the duplicate ones by checking structures that have almost the same energies after optimization, and select the ones within a 5 kcal/mol energy window under the small basis set. Therefore, the workflow would be:
- convert all structures in the
cluster.xyzto gaussian filescluster01.com,cluster02.com,cluster03.com… using B3LYP-D3(0)-PCM(your solvent)/6-31G(d) level of theory using the command of:#p Opt=(gdiis, recalc=40) rb3lyp/6-31g(d) em=gd3 scrf=(solvent=methanol) - Get the outputs and then read out the energies of your structures. This can be done by one simple line:
for a in *.log; do echo $a; cat $a | grep 'SCF Done' | tail -n 1 | awk '{print $5}'; done | xargs -n2This script will read all the .log files in the folder, extract the energy value after the very last “SCF Done,” and then echo the file name and the energy in Hartrees.
After getting the list of energies, keep the structures within a 5 kcal/mol window and optimize them with your target level of theory – this will save you a great deal of time by removing high-energy structures and duplicates while providing better initial guess structures!
- Write the optimization input files for the candidates that proceed to the next round with the new command:
#p Opt=(gdiis, recalc=40) rb3lyp/6-31+g(d,p) freq em=gd3 scrf=(solvent=methanol)Run and wait.
- fter you obtain the optimized structures, order them according to their free energies. The structures within an energy window of 3 kcal/mol will be those that we think are really contributing to the macroscopic NMR signal and the ones that advance to the final stage – NMR calculation!
NMR shielding tensor calculation
The shielding tensor calculation is straightforward from the aspect of a simple command in Gaussian. However, understanding gauge-including atomic orbitals (GIAOs) requires physical chemistry knowledge, which is omitted here. In Gaussian, you may use GIAO to compute the shielding tensor for each atom in a structure using:
#p nmr=giao mpw1pw91/6-311+g(2d,p) scrf=(iefpcm,solvent=methanol)
remember to change the solvent and level of theory to your task.
referenceing and scaling the calculation
The shielding tensors of each atom can be easily saved using the GaussView interface. You will see an “NMR” option light up in your result panel, and when you click it, you will see a graph with signals. Right-click, and you can save the numbers for all elements in a .txt file.
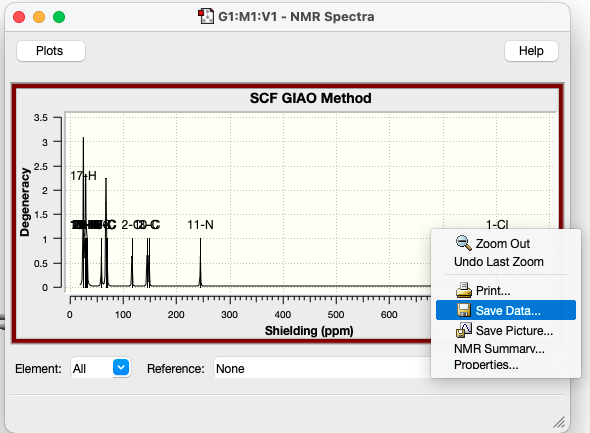
if you are using CHESHIRE:
Use the H and C scaling factor on the website to convert the numbers in text file to calculate the chemical shift(instruction of using factor is posted the CHESHIRE website).
if you are using TMS as reference:
To obtain the relative chemical shifts, follow these steps:
- Optimize the geometry of tetramethylsilane (TMS) and calculate the NMR shielding tensor using the same level of theory as your target molecules.
- Save the shielding tensor values for TMS.
- Compute the relative chemical shifts by subtracting the shielding tensor values of your target molecules from the corresponding values of TMS.
comparison
Spectral Simulation and Comparison The final step is to simulate the NMR spectrum based on the calculated chemical shifts and compare it with the experimental spectrum.
- Recognize Equivalent Atoms Identify
the equivalent atoms in the molecule, as they will have the same chemical shift. This can be done by analyzing the molecular symmetry and the chemical environment of each atom. Once the equivalent atoms are identified, calculate the average of their computed chemical shifts to obtain a single value for each set of equivalent atoms.
- Calculate Energy-Weighted NMR Signals
In many cases, molecules can exist in different conformations, each with its own set of chemical shifts. To account for the contribution of different conformers to the observed NMR spectrum, it is necessary to calculate the energy-weighted NMR signals. The equation would be:
\[\delta_{weighted} = \frac{\sum (\delta_i \times e^{-\Delta E_i / RT})}{\sum (e^{-\Delta E_i / RT})}\]- $δ_weighted$ is the energy-weighted chemical shift
- $δ_i$ is the chemical shift of the atom in conformer i
- $\Delta E_i$ is the relative energy of conformer i (in kcal/mol)
- $R$ is the gas constant (0.001987 kcal/mol/K)
- $T$ is the temperature (in Kelvin)
This equation essentially weights the contribution of each conformer based on its relative energy, with lower energy conformers contributing more to the final weighted chemical shift.
- Compare Estimated and Experimental NMR Signals To assess the agreement between the calculated and experimental NMR signals, several statistical measures can be used:
- Root-Mean-Square Deviation (RMSD) and Mean Absolute Error (MAE): measure the average deviation between the calculated and experimental chemical shifts. A lower RMSD/MAE value indicates better agreement between the calculated and experimental data.
- Coefficient of Determination: $R^2$ measures the linear correlation between the calculated and experimental chemical shifts. It ranges from 0 to 1, with 1 indicating a perfect linear correlation.
By comparing the calculated and experimental NMR signals using these statistical measures, you can assess the quality of your NMR calculations and determine if they provide reliable support for structure verification. This is particularly useful when you have several candidate molecules and you are unsure which one the NMR experiment is pointing to. In such cases, you can calculate the NMR signals for each candidate molecule and compare them with the experimental data. By evaluating the statistical measures, such as RMSD, MAE, and $R^2$, for each candidate, you can determine which structure has the best agreement with the experimental results.
After party
Why my computed result does not agree with experiment? Based on all the contents above, there are several reasons why your computed NMR results may not agree with the experimental data. Before doubting the structure itself, make sure that the calculation is well structured.